
Sequencing a genome is just the beginning. The real challenge lies in understanding what those billions of A's, T's, G's, and C's actually mean. For novel organisms without well-characterized relatives, genome annotation remains one of bioinformatics' most demanding tasks - traditionally requiring teams of experts clicking through screens for weeks.
GenomeAnnotator changes that. It accelerates annotation by combining homology, expression, and literature evidence, and continuously learns from researcher corrections to improve its predictions.
The Annotation Challenge
Genome annotation assigns biological functions to all genes in a genome using ab initio prediction, homology-based approaches, and structural definitions [1]. For novel organisms, this process faces significant hurdles.
Gene identification depends heavily on assembly quality, which is often limited for non-model organisms [2]. Without annotated genomes from evolutionarily close species, both prediction and validation become challenging. Some organizations maintain teams of over 100 curators just to ensure annotation quality - that's expensive.
From Sequence to Knowledge
Raw Genome (FASTA)
>gene_0001
ATGGCCCTGTTTACGGAGATCCCGTTA...
>gene_0002
TTTACGACCGGAAGCTTTGAACCTGAA...
>gene_0003
AACGTGACCTTAGGATCTGCCCGATTC...These sequences contain no inherent meaning - just nucleotides waiting to be decoded.
Sequence --> Knowledge
GenomeAnnotator transforms raw sequences into structured, actionable annotations:
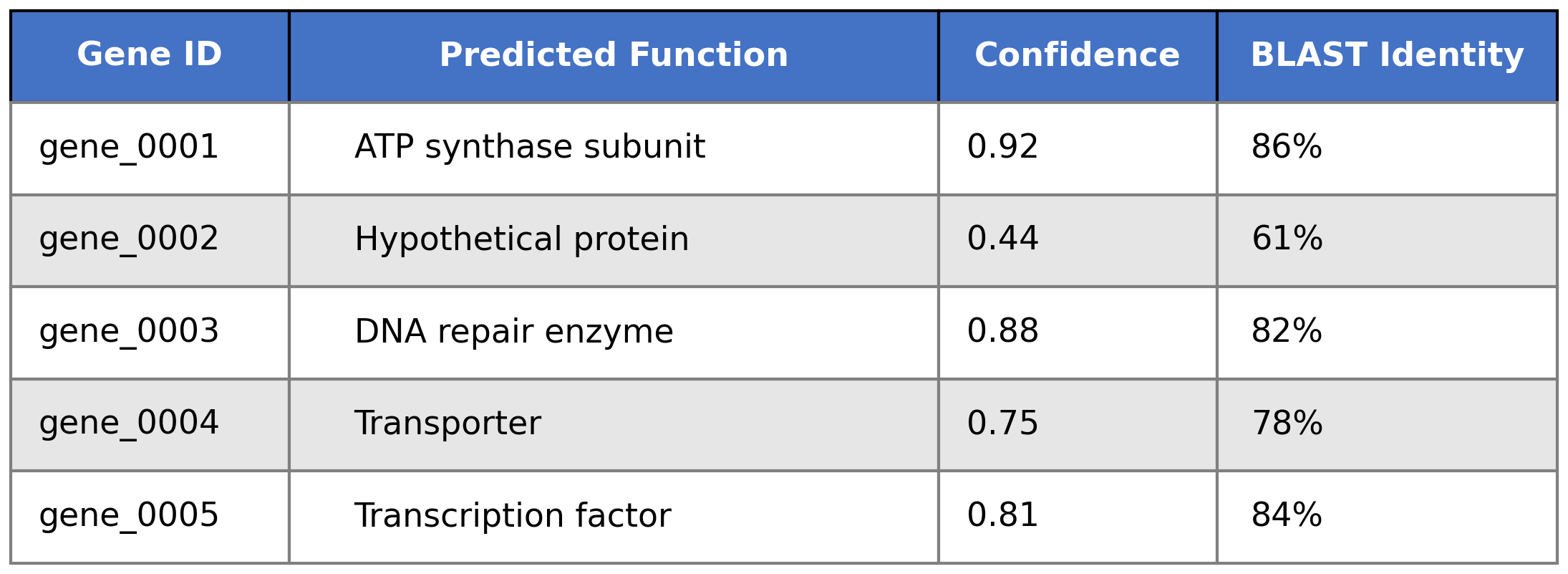
Each prediction includes confidence scores and BLAST identity, enabling researchers to prioritize validation efforts. High-confidence predictions (>0.85) can proceed directly to experimental work, while lower scores flag genes needing manual review.
Multi-Evidence Integration
GenomeAnnotator combines three complementary approaches:
Homology-Based Evidence: Comparing novel sequences against characterized proteins from related organisms. BLAST identity scores indicate match quality - higher identity (>80%) typically yields confident predictions [1].
Expression Evidence: RNA-seq data confirms genes are actually transcribed, identifies tissue-specific patterns, and validates gene boundaries [1].
Literature Evidence: Mining published research to link sequences to characterized protein families and surface experimental evidence for predicted functions.
The Learning Loop
What distinguishes GenomeAnnotator from static pipelines is continuous learning. When researchers correct a prediction - adjusting function, gene boundaries, or adding details - GenomeAnnotator incorporates this feedback to improve future predictions.
This addresses a key limitation: HMM models can annotate DNA sequences, but we have little knowledge of gene structures for newly sequenced genomes [1]. By learning from expert curation, GenomeAnnotator builds organism-specific models that outperform generic tools.
Deep Learning Integration
Early algorithms employed shallow learning with limited feature characterization [3]. Modern deep learning models are more capable and flexible - with appropriate training data, they automatically learn features with minimal human participation.
GenomeAnnotator leverages these advances to identify genomic elements, predict structures in low-homology regions, and integrate diverse evidence into unified confidence scores.
From Weeks to Hours
Traditional annotation requires running multiple tools separately, manually reconciling conflicts, searching literature, and curating gene models one by one. This takes weeks per genome.
GenomeAnnotator compresses this timeline by automating evidence integration while preserving expert oversight. Researchers focus on biological interpretation rather than computational housekeeping.
Applications
Novel Organism Characterization: Accelerate the path from raw sequence to biological understanding for new species.
Comparative Genomics: Consistent annotation across genomes enables meaningful cross-species comparisons.
Functional Studies: Reliable predictions guide experimental design for knockouts and protein characterization.
References
[1] Parca L, et al. "Review on the Computational Genome Annotation of Sequences Obtained by Next-Generation Sequencing." Briefings in Bioinformatics. 2020. https://pmc.ncbi.nlm.nih.gov/articles/PMC7565776/
[2] Chuguransky S, et al. "A benchmark study of ab initio gene prediction methods in diverse eukaryotic organisms." BMC Genomics. 2020. https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-020-6707-9
[3] Zhu Y, et al. "From tradition to innovation: conventional and deep learning frameworks in genome annotation." Briefings in Bioinformatics. 2024. https://academic.oup.com/bib/article/25/3/bbae138/7640738